
Notre objectif est d'identifier les gènes à l'origine des défauts des valves cardiaques et de comprendre les mécanismes qui contrôlent la formation des valves. Les études sur les modèles animaux ont permis d'identifier les principales étapes du développement des valves cardiaques ainsi que les principales voies de signalisation impliquées. Les valves semilunaires (aortique et pulmonaire) et atrioventriculaire (tricuspide et mitrale) se forment à partir de coussins endocardiques situés respectivement au niveau de la voie efférente et du canal atrio-ventriculaire. Au cours du développement, des cellules de l'endocarde bordant ces coussins vont les envahir et acquérir un phénotype de cellules mésenchymateuses (étape de transition endothélio-mésenchymateuse). Ces cellules donneront les cellules valvulaires interstitielles nécessaires à la synthèse de la matrice extracellulaire. Les valves continuent leur maturation après la naissance. Le flux sanguin permet l'acquisition de la structure trilaminaire des valves matures. La matrice extracellulaire des valves va se réorganiser de manière à former trois couches distinctes (la fibrosa, la spongiosa et la ventricularis) riche en fibres élastiques.
Les valvulopathies, congénitales ou acquises, sont des pathologies fréquentes des valves cardiaques. Environ 2% de la population adulte est porteuse d'une valvulopathie, insuffisance mitrale ou insuffisance aortique. Ces pathologies se définissent comme un dysfonctionnement d'une ou plusieurs valves cardiaques pouvant être consécutif à un défaut d'ouverture de la valve (sténose valvulaire) et/ou à un défaut de fermeture de la valve (insuffisance valvulaire). Le développement et le remodelage des valves cardiaques sont des processus complexes qui conduisent à une valve mature composée de matrice extracellulaire organisée en trois couches distinctes (collagènes, protéoglycanes et élastine). La composition de la matrice extracellulaire est nécessaire aux propriétés biomécaniques des valves cardiaques. Toutefois, les facteurs qui régulent l'homéostasie de ces composants sont très peu connus. Notre laboratoire vient de montrer que le facteur de transcription, Krox20 (aussi appelé Egr2 [early growth response 2]), est exprimé dans les valves cardiaques embryonnaires et adultes. De plus, la délétion génétique de Krox20 provoque une hyperplasie de la valve aortique, conduisant chez l'adulte à une régurgitation aortique (insuffisance aortique). Cette anomalie ressemble à une dégénérescence (myxoïde) de la valve aortique, comprenant un excès de protéoglycanes et une réduction des fibres de collagène, en particulier les collagènes de type I et III. Notre étude montre que Krox20 est nécessaire à la régulation de l'activité transcriptionnelle des gènes de collagènes fibrillaires, Col1a1 et Col3a1. Ainsi, nos résultats aident à mieux comprendre le mécanisme qui peut conduire à la maladie de la valve aortique. Odelin et al., 2014
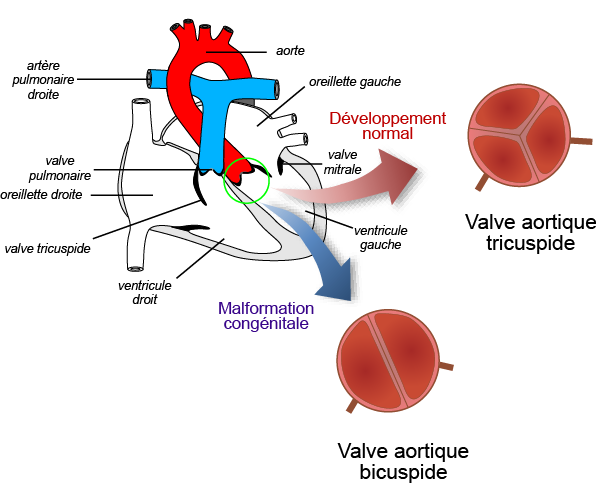
Les différentes études menées sur les modèles animaux ont permis de mieux comprendre le développement normal des valves (valvulogenèse). Ces structures se forment à partir de cellules présentes au niveau du cœur (cellules endothéliales) mais également à partir de cellules extracardiaques qui vont migrer de la partie dorsale du tube neural (appelées cellules cardiaques de la crête neurale) jusqu'au cœur pendant son développement. Notre équipe a identifié une sous population de cellule de la crête neurale qui contribue à la formation des bords libres des feuillets valvulaires mais également de leur zone d'insertion au niveau de l'aorte. Ces cellules sont caractérisées par l'expression du facteur de transcription, Krox20. Nes travaux ont montré que l'inactivation du gène Krox20 chez la souris conduit à une valvulopathie aortique. La valve aortique des souris mutantes pour ce gène sont alors hypertrophiées et bicuspide dans 30% des cas. Grâce à des outils génétiques permettant de suivre les cellules au cours du développement embryonnaire, l'équipe a pu montrer que l'inactivation de Krox20 conduit à l'augmentation du nombre de cellules cardiaques de la crête neurale qui contribuent normalement au bord libre des feuillets de la valve aortique. Cet excès de cellules envahissant les feuillets interfère avec le développement normal de la valve aortique ce qui conduit dans certain cas à une valve aortique bicuspide. Odelin et al., 2018
La bicuspidie aortique est une anomalie anatomique de la valve aortique qui présente deux feuillets au lieu de trois feuillets (coronaire droit, coronaire gauche et non coronaire). Il s'agit de l'anomalie cardiaque congénitale la plus fréquente. On estime qu'environ 2% de la population présente cette anomalie. Bien que hautement héréditaire, peu de mutations causales ont été identifiées chez les patients porteurs de cette anomalie. Notre étude décrit l'identification de variants indel rares dans le domaine histidine homopolymérique du facteur de transcription HOXA1 chez des patients ayant une bicuspidie aortique. L'analyse in vitro montre que la perturbation de ce motif entraîne une réduction significative de la demi-vie de la protéine HOXA1et une activité transcriptionnelle anormale de ce facteur. Chez le poisson zèbre, le ciblage de l'orthologue hoxa1a entraîne des défauts de la valve aortique. Des tests in vivo indiquent que les variants identifiés chez l'homme se comportent comme des dominants négatifs entraînant un développement anormal de la valve chez le poisson lorsqu'ils sont injectés à un stade précoce. Chez la souris, l'invalidation du gène Hoxa1 conduit à une bicuspidie aortique associée un très petit feuillet non coronaire rudimentaire. Notre étude montre également que les souris portant une mutation perturbant le motif poly-histidine présentent un phénotype similaire. Le traçage génétique chez les souris Hoxa1-/- révèle une réduction anormale des cellules dérivées de la crête neurale dans le feuillet valvulaire, qui est causée par un défaut migration précoce de ces cellules. En conclusion notre étude identifie des variants indel rares dans le motif poly-histidine de HOXA1 associés à la bicuspidie aortique. Odelin et al., 2023
Ces travaux ont été soutenus par l'AFM-Telethon, la FRM et la Fondation Lefoulon-Delalande
